Research
Our research covers a range of medicinal chemistry and chemical biology areas focused on malaria, TB, antimicrobial resistance, pancreatitis and filariasis. More recently we have also initiated medicinal chemistry programmes in the discovery of small molecule inhibitors of snake venom for the treatment of snake bite.
Malaria
Endoperoxide Antimalarials
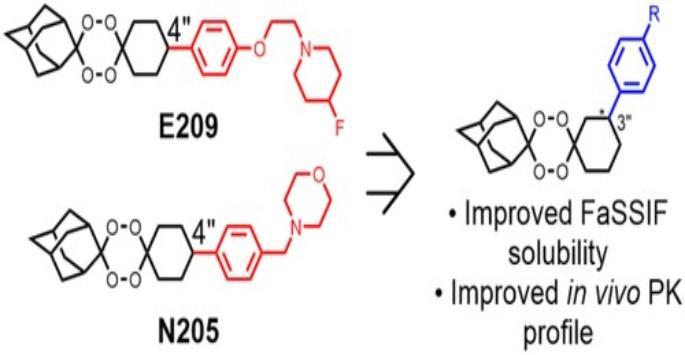
Synthetic endoperoxide antimalarials, such as 1,2,4-trioxolanes and 1,2,4,5-tetraoxanes, are promising successors for current front-line antimalarials, semisynthetic artemisinin derivatives. Previously at Liverpool, we have reported two second-generation tetraoxane antimalarials, E209 and N205, which showed potent in vitro antimalarial activity against artemisinin resistant PfK13 mutant strains of P. falciparum, and evidence for a single dose cure in the humanised SCID mouse model of P. falciparum infection. [1,2] However, limited solubility of second-generation analogues in biological-relevant (Fasted State Simulated Intestinal Fluid, FaSSIF) media represents a barrier in clinical development.
We recently reported synthesis of a series of nonlinear analogues of second-generation tetraoxane antimalarials to investigate reduced molecular symmetry on in vitro antimalarial activity and physicochemical properties. [3] While maintaining good antimalarial activity and metabolic stability, head-to-head comparison of linear and nonlinear counterparts showed up to 10-fold improvement in FaSSIF solubility for three of the four analogues studied. Pharmacokinetic studies in rats comparing a selected nonlinear analogue 14a and its parent N205 showed improvement on oral absorption and exposure in vivo with more than double the AUC and a significant increase in oral bioavailability (76% versus 41%). These findings provide support for further in vivo efficacy studies in preclinical animal species.
- O’Neill, P. M.; Amewu, R. K.; Charman, S. A. et al. A Tetraoxane-Based Antimalarial Drug Candidate That Overcomes PfK13-C580Y Dependent Artemisinin Resistance. Commun. 2017, 8, 15159
- O’ Neill, P. M.; Stocks, P. A.; Sabbani, S. et al. Synthesis and Profiling of Benzylmorpholine 1,2,4,5-Tetraoxane Analogue N205: Towards Tetraoxane Scaffolds with Potential for Single Dose Cure of Malaria. Med. Chem. 2018, 26 (11), 2996– 3005
- Woodley, C. M.; Nixon, G. L.; Basilico, N. et al. Enantioselective Synthesis and Profiling of Potent, Nonlinear Analogues of Antimalarial Tetraoxanes E209 and N205. ACS Med. Chem. Lett. 2021, 12, 1077– 1085
Dual Action P. falciparum Plasmepsin IX/X inhibitors
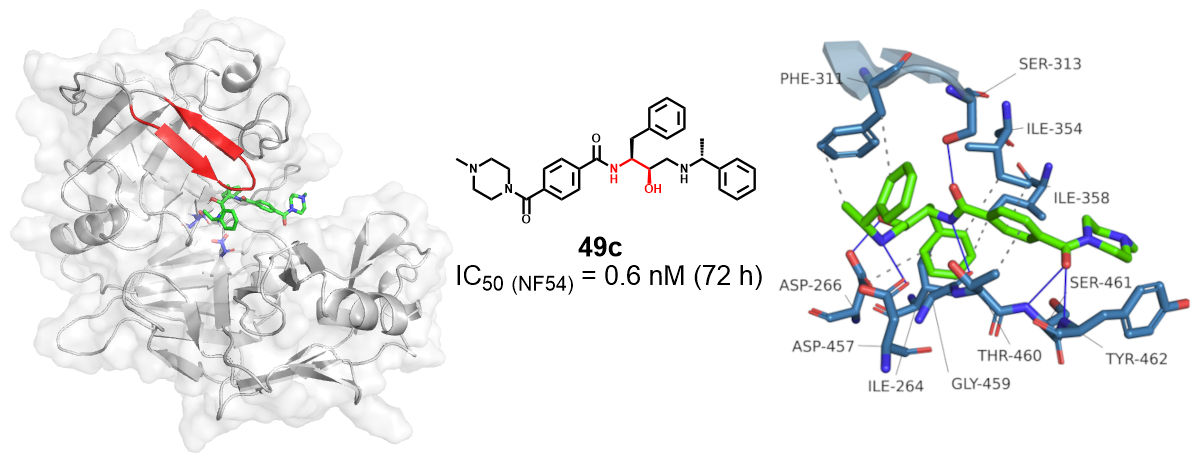
Plasmepsin IX and X (PMIX and PMX) are two isoforms within a series of ten aspartic acid proteases produced by P. falciparum. Both play a key role in parasite egress and invasion in the human host and mosquito vector. [1] PMX contributes to the maturation of serine protease PfSUB1 during the erythrocytic cycle, which is responsible for the parasitophorous vacuole membrane disruption and red blood cell membrane poration. It is also expressed when the gametocytes egress and ookinetes develop into oocysts. Meanwhile, PMIX is implicated in the maturation of rhoptry proteins such as PfRAP1, PfASP and PfRON3 during the invasion of red blood cells. In theory, PMIX/X dual inhibitors could be developed as mosquito, liver and blood stage antimalarials for both protection and treatment with a high barrier to parasite resistance. [2] A few compounds have been identified to inhibit both PMIX and PMX. Notably, 49c (also known as MMV1576861) with a hydroxyethylamine scaffold was reported as a promising multistage antimalarial lead compound. It exhibits extremely potent antimalarial activity after incubating with the parasite for 72 hours (IC50 = 0.6 nM) and reasonable metabolic stability in mouse liver microsomes (MLM = 75 μL/min/mg). [3] It also showed good selectivity over cathepsin D and is non-toxic to a control mammalian cell line. Furthermore, in vivo studies confirmed the efficacy of 49c in both P. falciparum and P. berghei mice models.
- Cheuka, P. M.; Dziwornu, G.; Okombo, J.; Chibale, K. Plasmepsin Inhibitors in Antimalarial Drug Discovery: Medicinal Chemistry and Target Validation (2000 to Present). Med. Chem. 2020, 63, 4445-4467.
- Pino, P.; Caldelari, R.; Mukherjee, B. et al. A multistage antimalarial targets the plasmepsins IX and X essential for invasion and egress. Science 2017, 358, 522-528.
- Ciana, C. L.; Siegrist, R.; Aissaoui, H. et al. Novel in vivo active anti-malarials based on a hydroxy-ethyl-amine scaffold. Med. Chem. Lett. 2013, 23, 658-662.
Vector Control: The role of chemosensory proteins in conferring pyrethroid resistance
Insecticide resistance is a major threat to global health and food security. Globally, vector borne diseases account for more than 17% of infectious disease annually, with over half the world's population currently at risk. Vector control relies heavily on the use of pesticides. The efficacy of insecticide control is exemplified by the success of malaria control programmes in Africa which have been heavily dependent on the use of pyrethroid insecticides in bednets. Between 2000 and 2015, insecticide treated bednets (ITNs) are estimated to have accounted for nearly 70 % of the reduction in number of malaria cases in Africa, contributing to halving the disease burden since the turn of the century. However, with this success comes a major challenge for the future sustainability of malaria control. As with intensive use of any drug or pesticide, the target organisms (in this case Anopheles mosquitoes), have developed widespread resistance to the chemicals used to control them (in this case pyrethroid insecticides), posing a critical threat to the future of malaria control. Understanding the mechanisms by which organisms develop resistance is critically.
We recently discovered a highly potent pyrethroid resistance mechanism in African Anopheles mosquitoes. An increase in the expression of a class of small proteins normally involved in chemical communications (and hence termed chemosensory proteins) in the legs of the mosquito acts as a sponge, absorbing the pyrethroid insecticides as it enters the mosquito via contact with the bednet. One specific member of this protein family, SAP2, is of key importance: mosquitoes that have elevated levels of SAP2 have a much greater chance of surviving pyrethroid exposure and, if we stop the mosquitoes producing this protein, this pyrethroid resistance largely disappears. This latter observation is remarkable as the mosquito populations tested contain additional well established resistance mechanisms including structural changes in the pyrethroid target site that reduce insecticide binding and elevated levels of enzymes that detoxify pyrethroids in the mosquito; the finding that silencing a single small protein can revert these mosquitoes to pyrethroid susceptibility opens up the exciting prospect that we may have found a way of blocking pyrethroid resistance in the mosquito, and potentially other pest species. In this project we are investigating exactly how increases in expression of this SAP2 protein plays such a pivotal role in pyrethroid resistance and are developing methods to break this resistance mechanism through cheminformatics aided screening and medicinal chemistry optimisation. This project is in collaboration with Hilary Ranson (LSTM, U.K.) and Victoria Ingham (Heidelberg University Hospital, Germany)
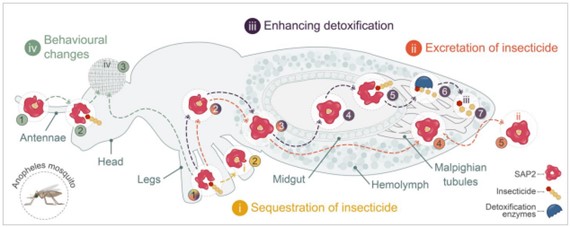
Figure 1: Potential mechanisms by which SAP2 confers resistance to insecticide
- Ingham, V.A., Anthousi, A., Douris, V. et al. A sensory appendage protein protects malaria vectors from pyrethroids. Nature 2020, 577, 376–380
Tuberculosis
A major failure of current tuberculosis (TB) therapies is that they predominantly target replicating Mycobacterium tuberculosis (Mtb) but are unable to sterilize slow growing (dormant) Mtb, leading to protracted treatment regimes and the development of drug resistance. We propose to generate a new drug against TB that is able to mitigate the shortcomings of current therapies, leading to improved treatment outcomes. Our strategy is to target the Mtb respiratory chain, specifically NADH:menaquinone oxidoreductase (ndh) (Figure 1). This target is essential for the survival of replicating, dormant and drug resistant Mtb, and it is absent in humans. Over the past 2 years we have successfully progressed from target validation/hit identification to the discovery of novel potent (nM) inhibitors of ndh with corresponding potent (nM) in vitro sterilization activity against replicating and dormant Mtb. We have received funding for a 1 million pound grant from the MRC to perform lead optimisation of our quinolone antitubercular compounds (Figure 2). The project is in collaboration with GSK, Tres Cantos, Madrid, Spain. This work is performed with Professor Steve Ward and Professor Giancarlo Biagini from the Liverpool School of Tropical Medicine.
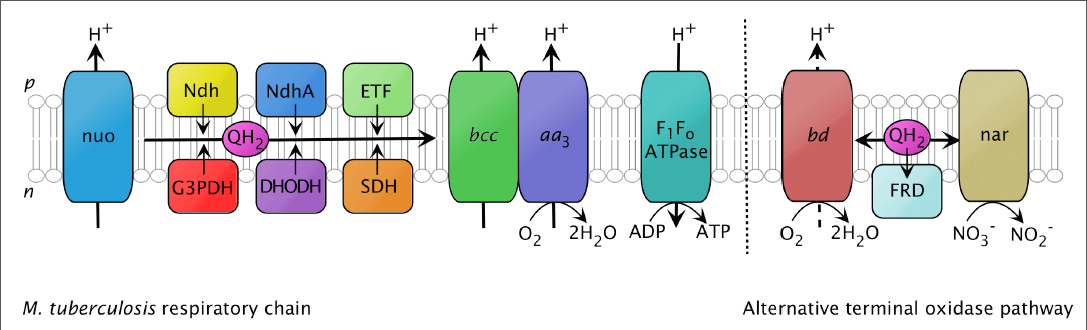
Figure 1: Cartoon representation of the electron transport chain in Mycobacterium tuberculosis. We are targeting the Ndh component.
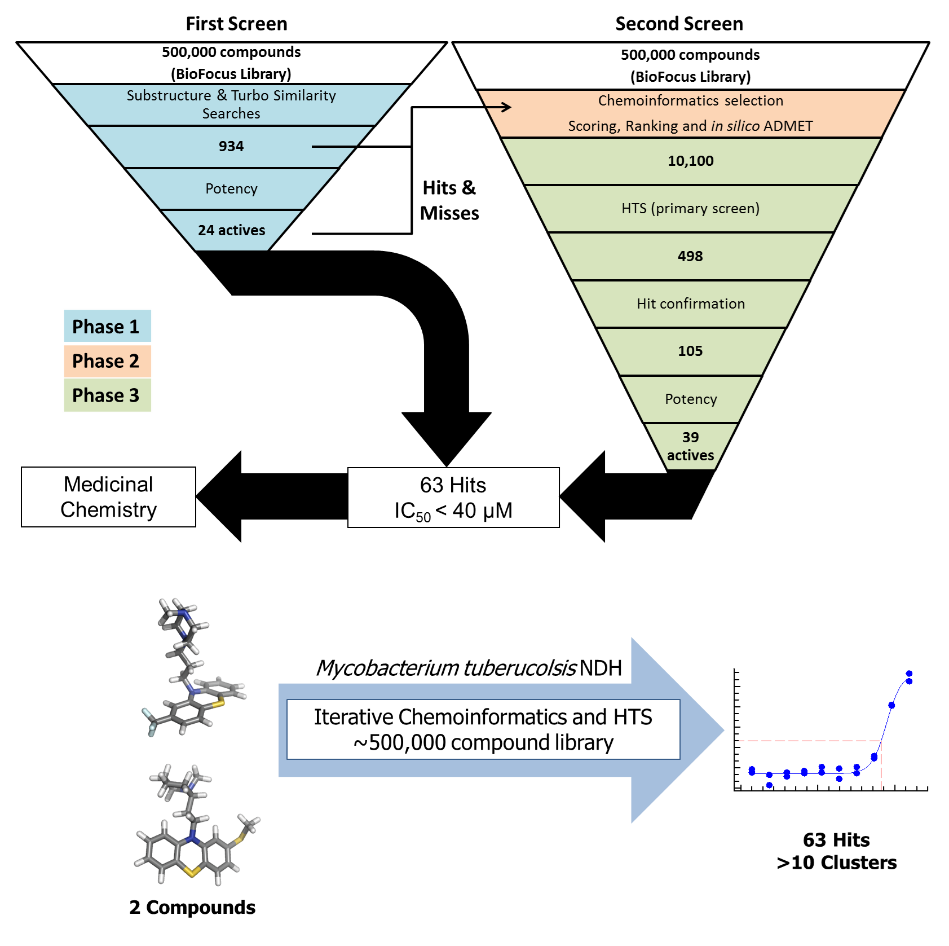
Figure 2: High throughput screening triage, informed by chemoinformatics, targeting the Ndh component of the electron transport chain of Mycobacterium tuberculosis. We have identified antituberculosis compounds which have now entered medicinal chemistry optimisation
Snake venom drug discovery
Discovery of Small Molecule Snake Venom Inhibitors
Snakebite envenoming is responsible for 81,000-138,000 deaths annually and 400,000 cases of long-term morbidity, making snakebite the most lethal neglected tropical disease (NTD) in the world. Currently the only treatment for snakebites are animal-derived antivenoms which have several issues with access in remote areas, specific storage requirements, and quality and standardisation of available antibodies. In our project we aim to develop an oral snakebite treatment as a standalone therapy or to be given as a rapid pre-hospital treatment to neutralise the effects of snakebite envenoming. We are investigating classes of inhibitors targeting the two main snake venom proteins; snake venom metalloproteinase (SVMP) and phospholipase A2 (PLA2). Since SVMP shares structural similarities with human matrix metalloproteinase (MMP), we aim to repurpose existing MMP inhibitors for SVMP inhibition.
Through high-throughput screening of more than 18000 compounds, we developed four series of SVMP inhibitors and three series of PLA2 inhibitors. Several analogues display potent activity against SVMP and moderate to excellent in vitro drug metabolism and pharmacokinetic (DMPK) profiles. To validate their effectiveness, we conducted in vivo experiments using snake envenomed mice models, testing the five most potent derivatives with optimal in vitro DMPK profiles. The selected derivatives also displayed good bioavailability and modest half-lives which encouraged us to focus on these main scaffolds for lead optimisation. Additionally, a few analogues displayed potent activity against PLA2 and demonstrated optimum in vitro DMPK profiles.
This project is supported by multiple cheminformatics and molecular modelling techniques, from homology modelling, molecular dynamics simulations in combination with molecular docking, high-throughput virtual screening, DFT methods and machine learning to gain novel insights into protein dynamics, target prioritisation, lead optimisation and drug-binding mode predictions. A major part of our computational work on snake venom is focused on exploring the chemical space through the expansion /enumeration of R-groups, to efficiently accommodate the active sites of SVMP and svPLA2, so as to develop novel, potent and less toxic small molecule inhibitors.
This work is funded by the Wellcome Trust, in collaboration with Prof. Nick Casewell at the Centre for Snakebite Research and Intervention at the Liverpool School of Tropical Medicine

1 R. H. Clare, S. R. Hall, R. N. Patel and N. R. Casewell, Trends Pharmacol. Sci., 2021, 42, 340–353.
2 D. P. Becker, et al , J. Med. Chem., 2010, 53, 6653–6680.
3 D. P. Becker, et al, J. Med. Chem., 2005, 48, 6713–6730.
Triple negative breast cancer (PROTAC)
Proteolysis targeting chimera (PROTAC) approaches in Medicinal Chemistry
PROTAC’s are heterobifunctional molecules that consist of 2 small chemical “warheads” joined by a linker (Figure 1A). They are constructed to bind to both the protein of interest (POI) and E3 ligase. The green E3 ligase ligand can be chosen to target specific E3 ligases, the linker can be varied to change the activity/stability of the compound and the POI ligand can be varied to target different POI, giving them huge scope to treat multiple diseases.
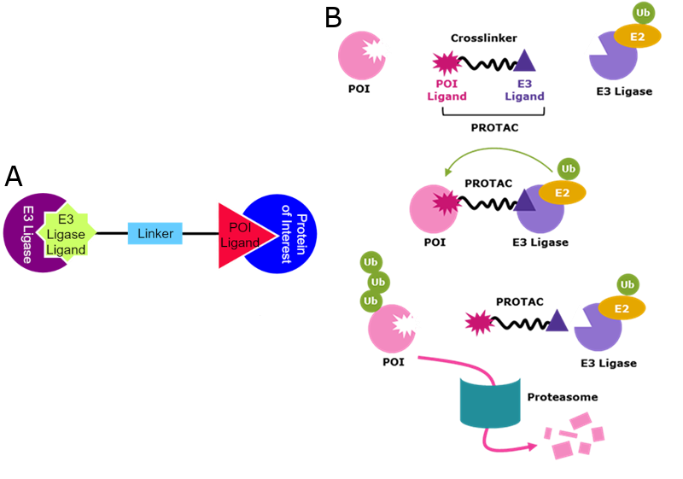
Figure 1: A) Components of a PROTAC compound. B) PROTACS and the UPS
The Ubiquitin/Proteasome System (UPS, Figure 1B) is a highly regulated mechanism of intracellular protein degradation and turnover. Through the concerted actions of a series of enzymes, proteins are marked for proteasomal degradation by being linked to the polypeptide co-factor, ubiquitin. The PROTAC recruits the POI to E3 ligase. Ubiquitin (Ub) is then added to POI Lys residues and the Ub-marked POI is degraded by the proteasome. Thus removing the target protein, rather than inhibiting it.
PROTAC Approaches to target S100A4 in the treatment of triple negative breast cancer
Breast cancer is the most common cancer in females worldwide, with 2.2 million cases in 2020. TNBC accounts for around 15-20% of breast cancer cases and is more likely to metastasise to other organs. S100 proteins are small, dimeric, calcium-dependent proteins involved in the regulation of cellular processes such as growth, motility and differentiation. S100 proteins are classified as metastasis inducing proteins (MIPs). They are the highest expressed MIP in TNBC. S100A4 binds to nonmuscle myosin IIA (NMIIA) and triggers a direct increase in cell migration and invasion – the principle steps in the formation of metastases. Genomic knockout of S100A4 significantly reduces the metastasis of cancer cells in mice. This providing a strong rational for targeting the destruction of S100A4 through a PROTAC approach. This led to the successful development of PROTAC RGC-01-05-18 (Figure 2). [1] This project was carried out in collaboration with Prof Philip Rudland (ISMIB, University of Liverpool).

Figure 2: Development of PROTAC RGC-01-05-18
- Ismail, T. et al, Targeted Destruction of S100A4 Inhibits Metastasis of Triple Negative Breast Cancer Cells. BIOMOLECULES, 2023, 13(7). doi:10.3390/biom13071099
Motor neurons disease

Above: members of the group working on the development of novel SOD1 degraders for the treatment of MND
Design and development of novel SOD1 degraders for the treatment of Motor Neurone Disease
Motor Neurone Disease (MND) is a rare condition that progressively damages parts of the nervous system eventually leading to death. Current treatment options are limited to two drugs that have limited benefit. Point mutations in SOD1 are the known cause for triggering MND in up to 20 per cent of patients who have a family history of the disease and around 5% of sporadic MND in the Western hemisphere, while in China they represent some 50% of familial disease cases. There is increasing evidence that toxicity of mutant superoxide dismutase-1 (SOD1) in MND is linked to its propensity to misfold and to aggregate. Degradation of these differently folded states of SOD1 has the potential to significantly improve patient outcomes. Through collaboration with Prof Samar Hasnian (ISMIB, University of Liverpool) two ligand-binding pockets in human SOD1 and its pathogenic mutants have been discovered and visualised via cocrystallography (Figure 1). [1,2] A small fragment based screen has identified hit fragments for both pockets that are now ready to be grown into potential inhibitors. Once initial inhibition and binding has been proven a Proteolysis-targeting chimera (PROTAC) approach will then be deployed to ultimately facilitate SOD-1 protein degradation.
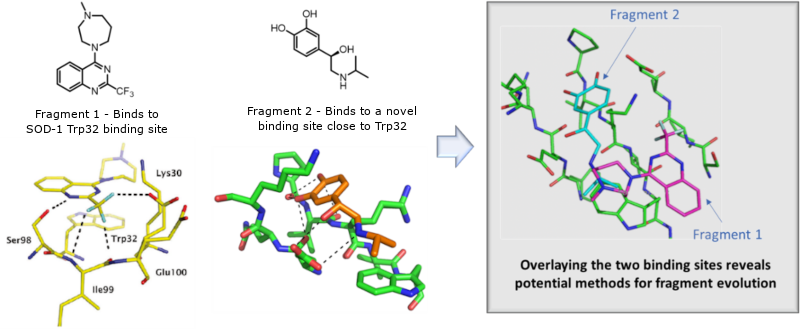
Figure 1: Fragment screening hits and potential for evolution and growth
- Antonyuk, S. et al. J Med.Chem 2010, 53, 1402–1406.
- Wright, G. et al. Nat. Commun., 2013, 4, 1758 | DOI: 10.1038/ncomms2750.
Antimicrobial peptides
Design and syntheses of highly potent teixobactin analogues
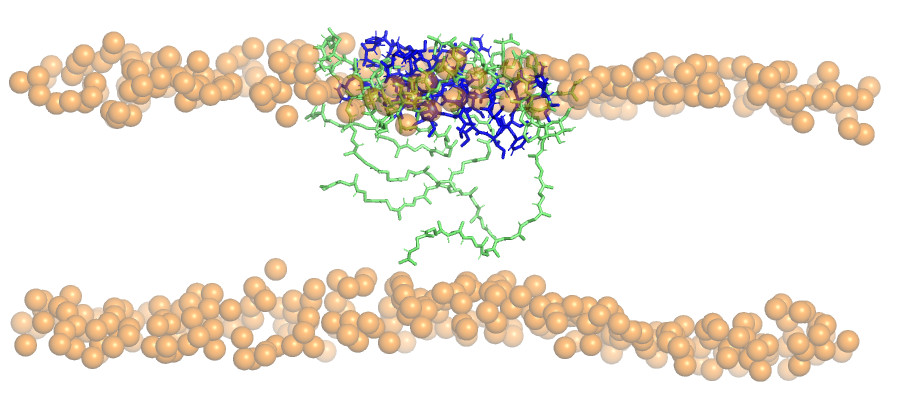
Figure 1: Snapshot from a molecular dynamics simulation of the tiexobactin-lipid II complex interacting with a DOPC lipid bilayer.
Most antibiotics used or under development for treatment suffer from resistance issues leading to treatment failures. For example, in 2019 nearly five million people lost their lives due to antibiotic resistance-associated infections and millions more live with poor quality of life due to treatment failures. Moreover, the development pipeline of innovative antimicrobials is nearly dry. As a group, “we aspire to bring new hope to improve and save lives currently lost due to AMR. We aim to achieve this by refreshing the antimicrobial pipeline with innovative molecules.”
For example, to address the developmental challenges such as safety and scalability of teixobactin, we have simplified teixobactins (depicted in Figure 1) to achieve good safety, scalability and efficacy. Teixobactin is labelled as a game changer as it kills the bacteria such as MRSA without detectable resistance by hiting multiple bacterial targets. We have successfully treated MRSA infections in several in vivo preclinical models.
- Parmar, A. et al, Design and syntheses of highly potent teixobactin analogues against Staphylococcus aureus, Methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococci (VRE) in vitro and in vivo. J. Med. Chem. 2018, 61(5) 2009–2017
Antimicrobial small molecules
Novel antibiotics developed from natural products
The global spread of the antimicrobial resistant pathogens is one of the biggest threats affecting the humankind in the 21st century. The most recent reviews estimated 4.95 million deaths attributed to antimicrobial resistance (AMR) in 2019, from which 1.27 million were deaths associated with bacterial AMR. Amongst the 6 leading bacterial pathogens for deaths associated with AMR, Staphylococcus aureus is responsible for one of the top two deadliest bacterial infections (> 100,000 deaths worldwide in 2019) and causes a large number of infections that require hospital treatments and has huge social and economical impacts.
18-beta-Glycyrrhetinic acid (GA, Figure 1) belongs to the pentacyclic triterpene family of natural products that can be modified to produce analogues with good potency against both sensitive and resistant S. aureus. Early reports from our group and others showed that GA derivatives have the potential to be further developed as novel antibiotics against Gram-positive bacteria. In this project, we use a semi-synthetic approach to derivatise GA, medicinal chemistry approaches to explore the structure activity relationship (SAR) and structure (DMPK) property relationship (SPR) of the GA scaffold, and microbiology, genomic and chemical proteomic experiments to investigate the modes of action/resistance of the antibacterial GA analogues.
This project is in collaboration with Adam Roberts (LSTM, U.K.) and Panpan Wu (Wuyi University, China)
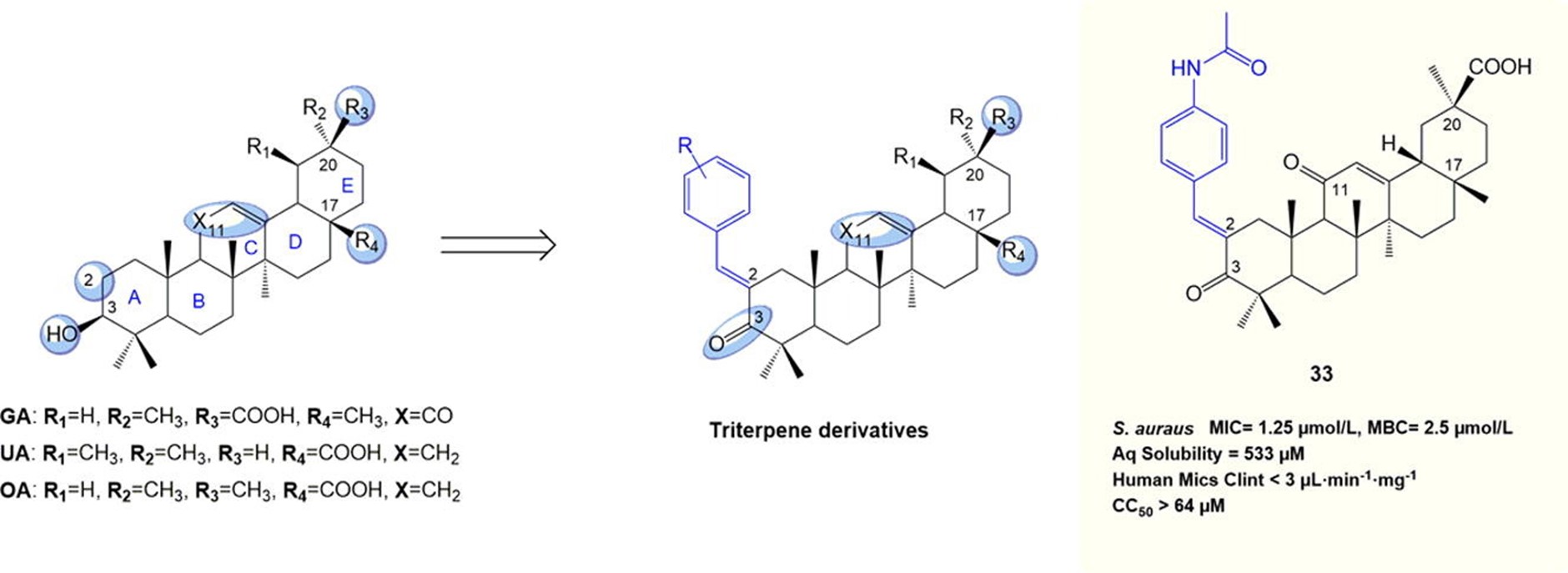
Figure 1: Development of the GA scaffold as antimicrobial agents
- Wu, P. et al., Synthesis and biological evaluation of pentacyclic triterpenoid derivatives as potential novel antibacterial agents. BIOORGANIC CHEMISTRY 2021, 109, 104692
Novel metallo-beta-lactamase inhibitors based on natural products
Antimicrobials underpin much of modern medicine, specifically for treating infectious disease and prophylactically for other procedures such as organ transplants, cancer chemotherapy and childbirth. The availability of antimicrobials allows the development of robust healthcare infrastructures supporting society in general. Antimicrobial susceptible bacteria are able to, often rapidly, evolve resistance to all currently used antimicrobials. In particular, carbapenem-resistant Gram-negative bacteria are a serious threat to global public health and they have recently been identified as “critical” priority pathogens for which new antibiotics are urgently required by the World Health Organisation.
Metallo-beta-lactamases (MBLs, Figure 2) are some of key protein enzymes that mediate the carbapenem-resistance in Gram-negative bacteria. In this project, potential inhibitors of MBLs, such as natural products reported in the literature or small molecules identified in biological screens will be investigated (through chemical modifications guided by rational medicinal chemistry approaches) as therapeutic agents to tackle the carbapenem-resistance in Gram-negative bacteria. This project is in collaboration with Adam Roberts (LSTM, U.K.) and Lei Zhang (Southern China University of Technology, China)
![NDM-1 enzyme displaying position of active site, with insert showing the coordination of zinc activating nucleophilic water. [B] Graphical representation of NDM-1 active site with corresponding x-ray crystal structure (PDB:3ZR9), showing how residues are positioned relative to the zinc ions.](/media/livacuk/chemistry/research/medicinalchemistrygroup/Dave_research2.jpg)
Figure 2: [A] NDM-1 enzyme displaying position of active site, with insert showing the coordination of zinc activating nucleophilic water. [B] Graphical representation of NDM-1 active site with corresponding x-ray crystal structure (PDB:3ZR9), showing how residues are positioned relative to the zinc ions.
Novel antibacterial macrocycles for treatment of resistant bacteria lung infections in people with cystic fibrosis
A library of macrocycle compounds was screened phenotypically against a multi-drug resistant cystic fibrosis (CF) isolate (Liverpool Epidemic Strain) of Pseudomonas aeruginosa. The screen identified molecules that act as antimicrobial potentiators for tobramycin and/or colistin, which are mainstays of antimicrobial chemotherapy for CF lung infections. One of the series, represented by a hit molecule coded Ndkg, is a multi-class antimicrobial potentiator, able to increase the efficiency of bacterial killing for both the aminoglycoside and the polymyxin (Figure 3). The hit series have no structural overlap with any known macrocyclic antibiotic classes.
In this ongoing project, we are optimising the Ndkg series, for which we have extensive in vitro and preliminary in vivo efficacy data. These preliminary data were collected with a strong focus on CF, including primary screens using a CF P. aeruginosa strain, and confirmation of hits was undertaken in a range of CF-relevant infection models.
Our aim is to develop macrocycle potentiators for co-formulation with existing, and widely used, antimicrobial agents that are delivered by an inhaled route. This would benefit people with CF by facilitating more efficacious antimicrobial chemotherapy, allowing lower doses of high-toxicity agents, such as colistin and tobramycin, to be used. This would reduce the incidence of the unwanted side-effects of long-term antimicrobial therapy and an inhaled method of delivery, would mean that potentiator use would not add to the onerous treatment regimen for people with CF. This project is in collaboration with Jo Fothergill (UoL) and Daniel Neill (University of Dundee)
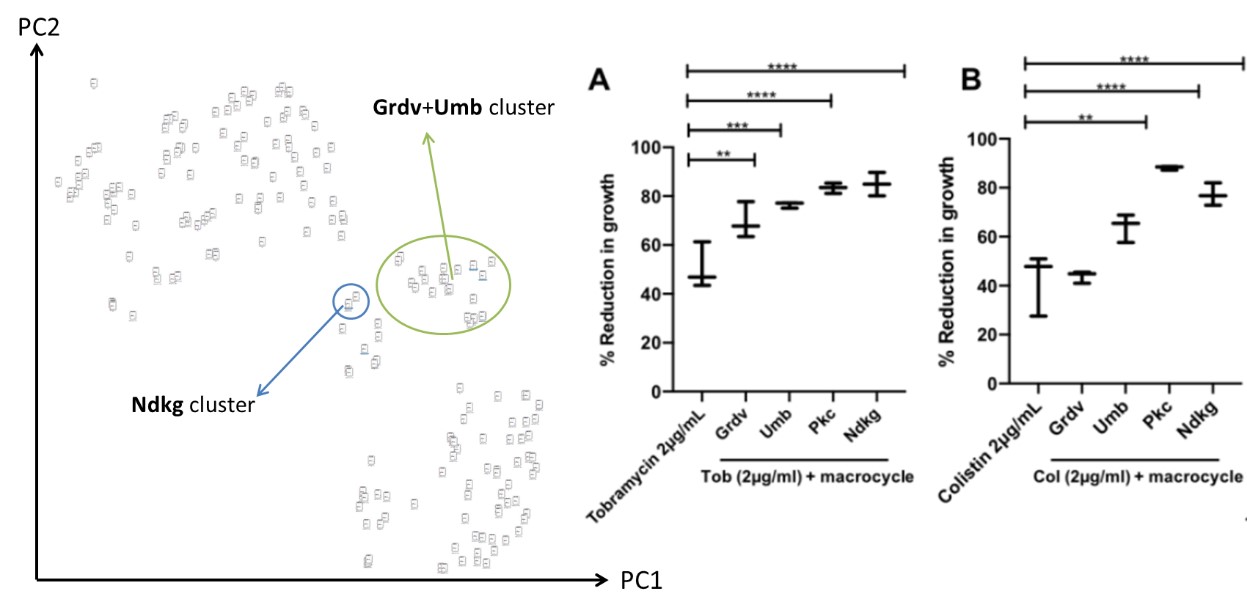
Figure 3: Macrocycle resistance-breaking hit clusters within screened library chemical space and sub-minimum inhibitory concentrations of tobramycin (A) or Colistin (B) +/- selected hits against Liverpool Epidemic Strain B65 (LESB65)..
Cryptococcus
Design, Synthesis and Biological Evaluation of β-tubulin binding benzimidazole like compounds for the treatment of Cryptococcus neoformans
Cryptococcus neoformans is yeast like fungus that occurs in both plants and animals. It is composed of two varieties; C. neoformans v. neoformans and C.n.v. grubii , as well as an extinct form C.n.v gattii.[1] In most cases it causes an infection of the lungs, however it can cause fungal meningitis and encephalitis, which is particularly problematic as a secondary infection in immunosuppressed patients, such as those suffering from AIDS.[2] It is estimated that there are 1 million cases of cryptococcal meningitis worldwide each year, of which 625,000 result in death.[3]
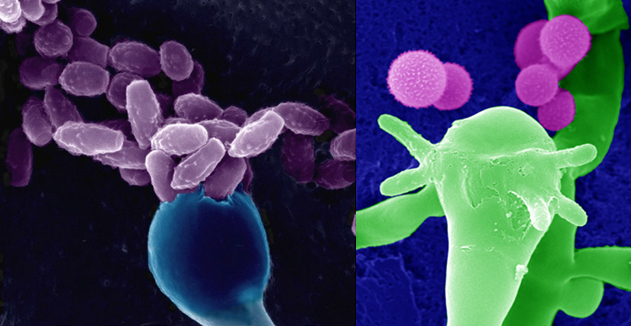
Figure 1: Scanning Electron Micrograph of the Cryptococcus neoformans yeast producing spores.[4]
Current treatments for the disease (amphotericin B, flucytosine and fluconazole) are associated with several issues including toxicity, poor activity and supply problems.[5] Evidently, new compounds with improved Drug Metabolism and Pharmacokinetic (DMPK) properties need to be generated for use in the treatment of cryptococcal infections.
The benzimidazole class of compounds has been historically used to treat helminth infections in humans and includes compounds such as albendazole and flubendazole. However, it has been noted that they also show good in vitro potency against C. neoformans. Benzimidazoles bind to the β-tubulin subunit of microtubules and cause disruption of the mitosis pathway. Furthermore, characterisation of the β-tubulin genes of Cryptococcus neoformans has been achieved and it was found TUB1 is the primary target of the benzimidazole class of compounds.[6]
The medicinal chemistry aspect of this project will involve the exploration of the SAR of both flubendazole and albendazole, in order to improve DMPK properties and the safety profile. This will also utilise predictive models to help target the design of albendazole and flubendazole analogues. Any analogues that are produced will be assessed for in vitro activity, in collaboration with Prof. William Hope (University of Liverpool, Institute of Translational Medicine), and will allow for redesign of the compounds to understand the SAR and improve DMPK properties throughout the project. Selected compounds demonstrating suitable potency and DMPK properties will undergo in vivo PK/PD experiments in the murine model of C. neoformans infection.

Figure 2: Strategy for exploration of the SAR of flubendazole and albendazole
- K. J. Kwon-Chung and J. E. Bennett, American Journal of Epidemiology, 1984, 120, 123-130
- J. N. Steenbergen and A. Casadevall, Journal of Clinical Microbiology, 2000, 38, 1974-1976.
- CDC, Centre for Disease Control and Prevention, 2014.
- C. Xue and K. Carroll, Live Science, 2013.
- S. J. Antony, A. Patel and J. Leonard, Journal of the National Medical Association, 1997, 89, 694-695.
- M. C. Cruz, M. S. Bartlett and T. D. Edlind, Antimicrobial Agents and Chemotherapy, 1994, 38, 378-380.
Cyclophilins as a drug target
Lead Series Development & Optimisation of a New Drug Against Acute Pancreatitis
The mitochondrial permeability transition pore (MPTP) plays an important role in damage-induced cell death, and agents inhibiting this pore may have a therapeutic potential for treating human conditions such as ischemia/reperfusion injury, trauma, and neurodegenerative diseases.
Opening of the MPTP causes mitochondrial dysfunction and necrosis in acute pancreatitis, a condition without specific drug treatment. Cyclophilin D (CypD) is a mitochondrial matrix peptidyl-prolyl isomerase that regulates the MPTP and is a drug target for acute pancreatits. We have synthesized urea-based small molecule inhibitors of cyclophilins and tested them against CypD using binding and isomerase activity assays. Our most potent compound to date shows enzymatic inhibition in the nanomolar range and is undergoing further optimisation as a novel small molecule agent against acute pancreatitis (Figure 1).[1]
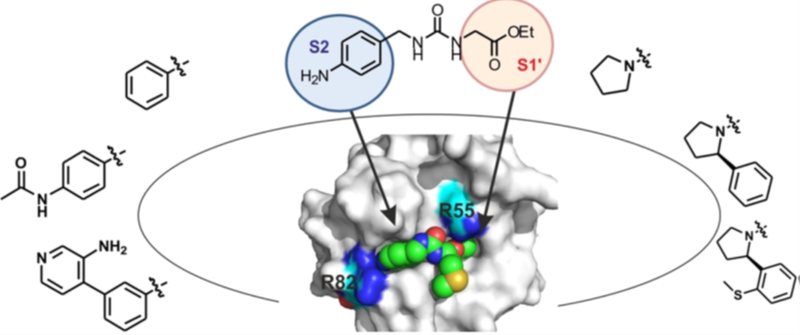
Figure 1: Design of potent inhibitors of Cyclophilin D
This work is in collaboration with Prof. Robert Sutton from the Institute of Translational Medicine and Prof. Lu-Yun Lian from the Instute of Integrative Biology.
1. Shore, E. R., Awais, M., Kershaw, N. M., Gibson, R. R., Pandalaneni, S., Latawiec, D., Sutton, R. et. al. (2016). Small Molecule Inhibitors of Cyclophilin D to Protect Mitochondrial Function as a Potential Treatment for Acute Pancreatitis. J. Med. Chem., 59(6), 2596-2611.
Filariasis
AWZ1066S, a highly specific anti- Wolbachia drug candidate for a short-course treatment of filariasis
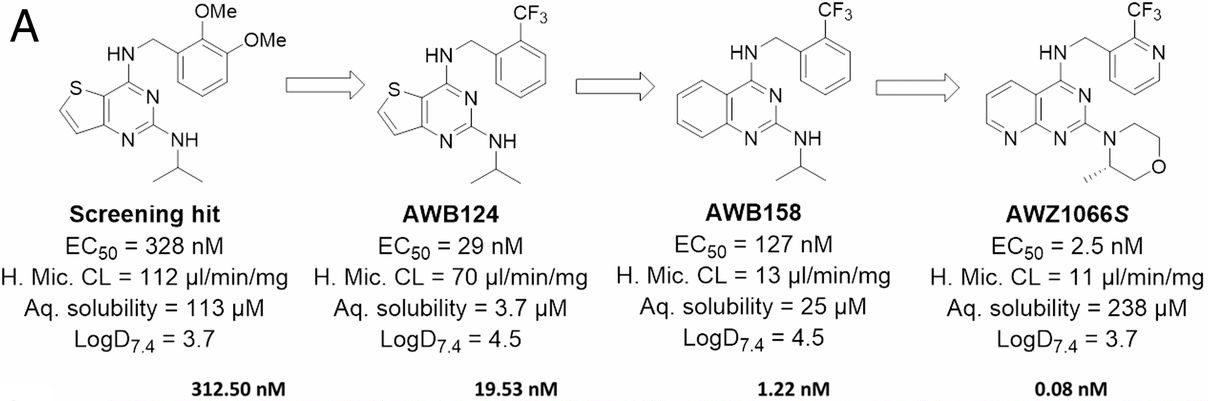
Figure 1: Development of AWZ1066s from the initial high-throughput phenotypic screening.
Onchocerciasis and lymphatic filariasis are two neglected tropical diseases that together affect ∼157 million people and inflict severe disability. Both diseases are caused by parasitic filarial nematodes with elimination efforts constrained by the lack of a safe drug that can kill the adult filaria (macrofilaricide). Previous proof-of-concept human trials have demonstrated that depleting >90% of the essential nematode endosymbiont bacterium, Wolbachia, using antibiotics, can lead to permanent sterilization of adult female parasites and a safe macrofilaricidal outcome. AWZ1066S is a highly specific anti-Wolbachia candidate selected through a lead optimization program focused on balancing efficacy, safety and drug metabolism/pharmacokinetic (DMPK) features of a thienopyrimidine/quinazoline scaffold derived from phenotypic screening (Figure 1). AWZ1066S shows superior efficacy to existing anti-Wolbachia therapies in validated preclinical models of infection and has DMPK characteristics that are compatible with a short therapeutic regimen of 7 days or less. This candidate molecule is well-positioned for onward development and has the potential to make a significant impact on communities affected by filariasis.
- Hong WD, Benayoud F, Nixon GL, Ford L et al. AWZ1066S, a highly specific anti-Wolbachia drug candidate for a short-course treatment of filariasis. Proc Natl Acad Sci U S A. 2019 ;116(4):1414-1419.
Medicinal Chemistry of Anti-Wolbachia Drugs (LSTM, Bill and Melinda Gates Foundation)
Our drug discovery and development portfolio also includes a Gates funded programme on novel anti-Wolbachia drugs for lymphatic filariasis. This programme, in collaboration with AstraZeneca, has revealed over ten new antibacterial chemotypes primed for medicinal chemistry optimization and we are actively engaged in hit to lead optimization.
This work is in collaboration with Professor Steve Ward and Professor Mark Taylor at the Liverpool School of Tropical Medicine and is part of the AWOL consortium.
Molecular modelling
Molecular modelling is used within the medicinal chemistry group i) to understand chemical systems more completely, ii) propose new molecular candidates for synthesis and testing and iii) predict properties of new molecules delivering novel research and training. This includes using chemoinformatics and both structure- and ligand- based modelling approaches applied to medicinal chemistry challenges. We use techniques such as Molecular Mechanics, Molecular Dynamics, Quantum Mechanics, Machine Learning, Library Design, 2D/3D QSAR, ADMET, De novo design, Docking, Pharmacophore, Molecular alignment, Structural Biology and Data pipelining. An applied example of our cheminformatic approach is provided in Figure 1, where from a library of 1.3 million compounds screened by AstraZeneca, we were able to characterise and prioritise multiple hits with excellent in vitro activity against the bacteria Wolbachia. [1] These compounds provide excellent starting points for optimisation and are part of the AWOL antifilarial drug discovery project with the Liverpool School of Tropical Medicine.
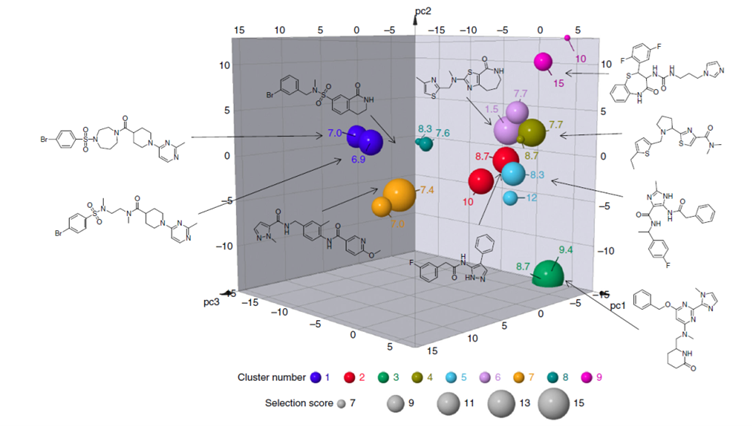
Figure 1: Eighteen selected fast acting macrofilaricides hits represented in chemical space. The first three principal components (PC1–3) together account for 31.6% of the overall variance in the data set. The chemotypes show clustering in chemical space. The size of each data point is proportional to the Selection Score. The label for each point is the ligand efficiency-dependent lipophilicity index (LELP) with values <10 considered good. A selection of the best compound structures is shown (large spheres) in a 3D plot produced by Datawarrior. The cluster number is indicated by colour: cluster 1 (dark blue sphere), 2 (red sphere), 3 (green sphere), 4 (dark gold sphere), 5 (light blue sphere), 6 (lilac sphere), 7 (orange sphere), 8 (teal sphere), 9 (pink sphere).
- Clare, R.H., Bardelle, C., Harper, P. et al. Industrial scale high-throughput screening delivers multiple fast acting macrofilaricides. Nat Commun 10, 11 (2019)
Chemical biology
Hybrid antibiotic probes for exploring collateral sensitivity networks in a panel of resistant clinical E. coli isolates
Reproducible collateral-sensitivity (CS) networks in Escherichia coli was discovered recently. CS occurs when sensitivity to one drug increases upon the development of resistance to another drug. Hybridisation of two antibiotics can lead to increased therapeutic efficacy; however the choice of antibiotics has never been informed by CS. This project aims to investigate novel hybrid (fluoroquinolone and aminoglycoside) antibiotics against E. coli, and determine if CS between these two classes of antibiotics will lead to a reduction in resistance development and persistence through a hybridisation strategy. This project is in collaboration with Adam Roberts (LSTM, U.K.).
- Podnecky, N.L., Fredheim, E.G.A., Kloos, J. et al. Conserved collateral antibiotic susceptibility networks in diverse clinical strains of Escherichia coli. Nat Commun 2018, 9, 3673
Chemical biology (MRC, BBSRC, EU)
Areas include, The biomimetic Fe(II) chemistry and EPR spectroscopy of new peroxide containing antimalarials; Mechanism based design of novel endoperoxide protease inhibitor pro-drugs; Application of proteomic techniques to identifying (i) the cellular targets of the artemisinin using biotinylated probe molecules (ii) P450s responsible for resistance to pyrethroid insecticides (Figure 1) using Click methodologies and rational probe design and synthesis.[1]
This work is in collaboration with Dr Mark Paine and Dr Hanafy Ismail at the Liverpool School of Tropical Medicine.
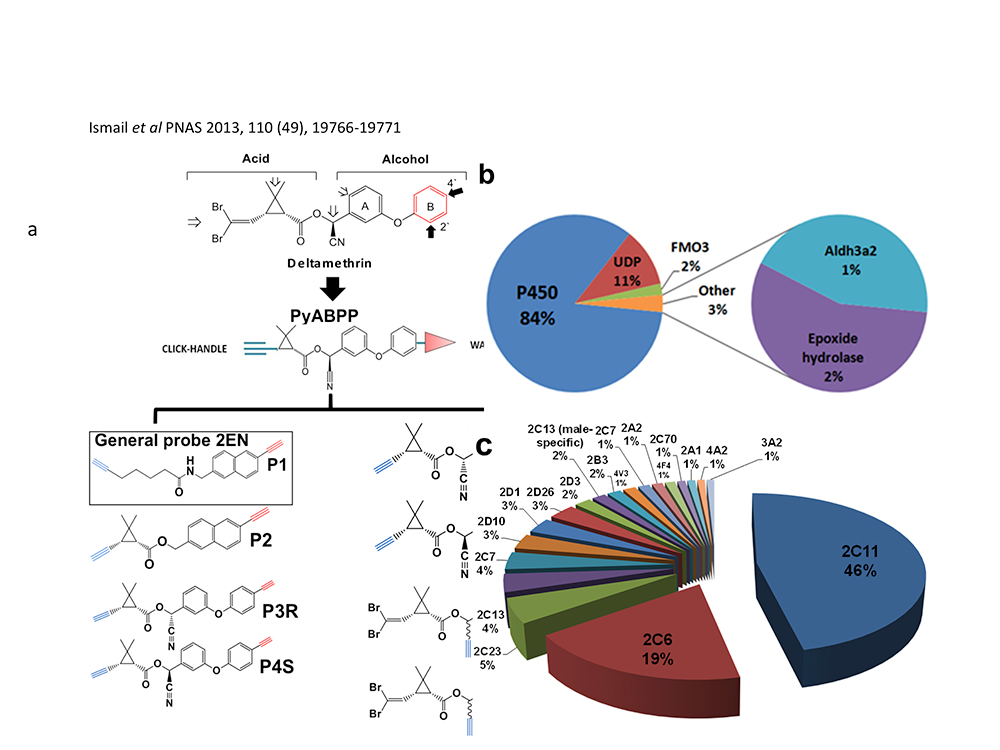
Figure 1: Chemical biology of pyrethroid insecticides.
1. Ismail, H. M.; O'Neill, P. Met al. Pyrethroid activity-based probes for profiling cytochrome P450 activities associated with insecticide interactions. Proc Nat Acad Sci USA 2013, 110, 19766-19771.