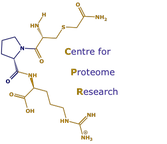
Derivatization for proteomics
Brancia, F.L., Butt, A., Beynon, R.J., Hubbard, S.J., Gaskell, S.J. & Oliver, S.G. (2001) A combination of chemical derivatisation and improved bioinformatic tools optimises protein identification for proteomics. Electrophoresis, 22, 552-559 [PUBMED] [PDF]
The identification of individual protein species within an organism's proteome has been optimised by increasing the information produced from mass spectral analysis through the chemical derivatisation of tryptic peptides and the development of new software tools. Peptide fragments are subjected to two forms of derivatisation. First, lysine residues are converted to homoarginine moieties by guanidination. This procedure has two advantages, first, it usually identifies the C-terminal amino acid of the tryptic peptide and also greatly increases the total information content of the mass spectrum by improving the signal response of C-terminal lysine fragments. Second, an Edman-type phenylthiocarbamoyl (PTC) modification is carried out on the N-terminal amino acid. The renders the first peptide bond highly susceptible to cleavage during mass spectrometry (MS) analysis and consequently allows the ready identification of the N-terminal residue. The utility of the procedure has been demonstrated by developing novel bioinformatic tools to exploit the additional mass spectral data in the identification of proteome proteins from the yeast Saccharomyces cerevisiae. With this combination of novel chemistry and bioinformatics, it should be possible to identify unambiguously any yeast protein spot or band from either two-dimensional or one-dimensional electropheretograms.
The identification of individual protein species within an organism's proteome has been optimised by increasing the information produced from mass spectral analysis through the chemical derivatisation of tryptic peptides and the development of new software tools. Peptide fragments are subjected to two forms of derivatisation. First, lysine residues are converted to homoarginine moieties by guanidination. This procedure has two advantages, first, it usually identifies the C-terminal amino acid of the tryptic peptide and also greatly increases the total information content of the mass spectrum by improving the signal response of C-terminal lysine fragments. Second, an Edman-type phenylthiocarbamoyl (PTC) modification is carried out on the N-terminal amino acid. The renders the first peptide bond highly susceptible to cleavage during mass spectrometry (MS) analysis and consequently allows the ready identification of the N-terminal residue. The utility of the procedure has been demonstrated by developing novel bioinformatic tools to exploit the additional mass spectral data in the identification of proteome proteins from the yeast Saccharomyces cerevisiae. With this combination of novel chemistry and bioinformatics, it should be possible to identify unambiguously any yeast protein spot or band from either two-dimensional or one-dimensional electropheretograms.