Until recently, many people had thought that the interaction potentials for a molecule-surface system did not vary much from one surface site to another when the surface being considered was metallic. The reasoning was that the electrons on a metal surface spread out, i.e. they (at least the conduction electrons) are delocalised, and so the "electronic surface" is smoother than the "real one", as illustrated below.
 |
A close-packed metal surface is shown from above on the left. When viewed side-on, right, you can see how the electrons (shown by red contours indicating constant density) spill over and fill in the bridge sites between the atoms. |  |
Since it is the electrons that determine the bonding, and so therefore the
bonding energy (i.e. the potential energy surface), then this must also be quite
a smooth function of position across the surface, or so goes the argument. This
seems to work reasonably for He interacting with a surface, but this is not
really a chemical interaction. For molecules, where bonds break and re-form (or
try to) this does not hold.
Here's an example of what I mean: the following two elbow potentials have been
computed for hydrogen molecules (H2) interacting with the (111)
surface of Cu for different sites, but with the molecule in the same azimuthal
orientation, i.e. to get from one to the other you just slide the molecule
across the surface.
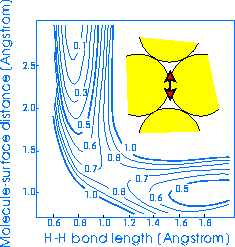
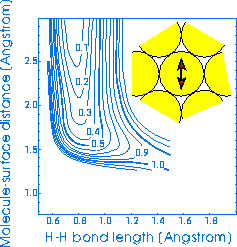
These potentials were calculated by
Bjørk Hammer
in Denmark, using a slab-supercell geometry to represent the surface. The
energies on the countours are in eV.
Consider the left-hand one first; this is the PES for dissociation from the
bridge site (the location of the molecular centre of mass) into the hollow site
(where the two atoms end up). This is the most favoured dissociation route, and
in fact the 'true' barrier height is lower than that obtained in this
calculation. Note that the elbow bends round very smoothly.
Compare it now
to the right-hand PES (for dissociation from on top of a surface atom with the H
atoms going into the hollow-site again). Notice that the atop-hollow PES has a
higher barrier, which occurs further from the surface and at greater bond-length
(these two actually cancel out somewhat), and that it bends around much more
sharply.
OK, so when the molecule comes down on different sites the PES looks
different.
But this is H2 /Cu(111) !! This is a paradigm of
activated dissociation dynamics which exhibits
normal energy scaling !!
So in other words, even though the different sites have very different
interaction PESs with different barrier heights, the dissociation still appears
as if the surface is flat.
To see how this comes about, we have to look at the PESs above in more
detail. The key point is that the barrier can change in two ways as the
molecule is moved across the surface; it can change its energy, e.g. from 0.73
eV to 0.96 eV, or it can change its location. We can see both of these in the
PES above. The atop-hollow dissociation route (on the right) has a barrier
which is energetically higher, and occurs at a greater distance from the surface
and at longer H-H bond length.
These different ways we can change the barrier (there are others which we
shall indicate below) give rise to different types of
corrugation of the PES.
The two basic types we call ENERGETIC
corrugation and GEOMETRIC
corrugation.
These two corrugations produce opposite and competing effects when the angle
of incidence is varied. If these are suitably balanced,
then normal energy
scaling results, as we shall show in the following pages.
Why does it appear flat in the dissociation ?
©G.R. Darling
<<Return to previous page
Return to index.
Return to my homepage.
Comments to
darling@ssci.liv.ac.uk