
Professor Dan Rigden BA, PhD
Professor of Protein Bioinformatics Biochemistry, Cell and Systems Biology
- +44 (0)151 795 4467
- Work email Drigden@liverpool.ac.uk
Research
Linking structural bioinformatics to software for experimental structural biology
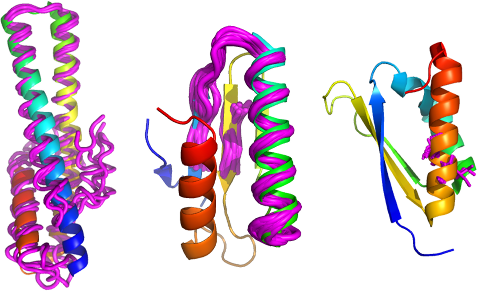
Structural bioinformatics offers much to X-ray crystallographers and Cryo-EM researchers alike. We have developed a variety of CCP4 pipelines to solve crystal structures by Molecular Replacement. We are also applying similar methods to expedite Cryo-EM map interpretation and to validate the results of both crystallography and Cryo-EM.
Protein structure modelling
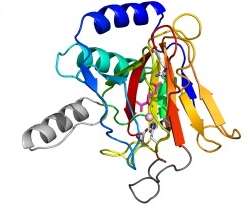
I have a broad interest in prediction of protein structures and complexes, especially with the latest Deep Learning-based methods like AlphaFold 2. This feeds directly into Molecular Replacement (above), often helps predict function (below) but has many other applications including interpreting the consequences of single nucleotide polymorphisms.
Protein function prediction
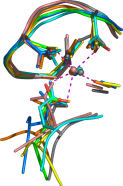
I am interested in using diverse bioinformatics methods to shed light on function of unannotated sequences - 'hypothetical proteins', Domains of Unknown Function' and the like. This often starts by using models for structure-based function prediction but also requires input from, for example, gene context, domain architectures, and structural bioinformatics.
Research Grants
Discovering the Origin of Vascular Aging Amyloid Protein Medin
NATIONAL INSTITUTES OF HEALTH (USA)
May 2022 - May 2024
Dissecting cell surface protein diversity to enhance leptospiral vaccine efficacy.
BIOTECHNOLOGY & BIOLOGICAL SCIENCE RESEARCH COUNCIL
September 2022 - December 2026
Exploiting the opportunities of new generation, high accuracy structure predictions for MR and more
SCIENCE AND TECHNOLOGY FACILITIES COUNCIL
January 2022 - December 2023
Extending the reach of Molecular Replacement with search model ensembles derived from new-generation protein models
SCIENCE AND TECHNOLOGY FACILITIES COUNCIL
January 2020 - December 2021
CCP4 Advanced integrated approaches to macromolecular structure determination
BIOTECHNOLOGY & BIOLOGICAL SCIENCE RESEARCH COUNCIL
October 2019 - September 2023
Establishing the genetic basis of symbiosis in an insect host
BIOTECHNOLOGY & BIOLOGICAL SCIENCE RESEARCH COUNCIL
June 2019 - January 2023
Studentship Agreement between University of Liverpool and Diamond Light Source Ltd
DIAMOND LIGHT SOURCE LTD (UK)
October 2018 - September 2022
Facilitating access to advanced Molecular Replacement pipelines via CCP4i2, CCP4 online and CCP4cloud
SCIENCE AND TECHNOLOGY FACILITIES COUNCIL
July 2017 - June 2019
Identification of novel double-stranded RNA elements in developing antibiotic resistance in the agricultural environment
NATURAL ENVIRONMENT RESEARCH COUNCIL
April 2016 - March 2018
Identification and evaluation of algal sulphatransferases for enzymatic polysaccharide modifications
BIOTECHNOLOGY & BIOLOGICAL SCIENCE RESEARCH COUNCIL
July 2017 - December 2017
Wellcome Trust Vacationship Miss Kiani A Jeacock
WELLCOME TRUST (UK)
June 2015 - August 2015
A Simkin Studentship
SOCIETE CIVILE SYNCHROTRON SOLEIL (FRANCE)
October 2015 - September 2019
Molecular function in post-genome biology (MOLFUN).
EUROPEAN COMMISSION
October 2006 - September 2010
The genotypic and phenotypic impacts of Shiga toxin encoding bacteriophage interactions with their host cells: consequences for food borne zoonoses
BIOTECHNOLOGY & BIOLOGICAL SCIENCE RESEARCH COUNCIL
August 2011 - November 2014
Dealkylation of plant sterols during utilization in invertebrates
LEVERHULME TRUST (UK)
May 2007 - May 2009
Tools for motif recognition in fungi
BIOTECHNOLOGY & BIOLOGICAL SCIENCE RESEARCH COUNCIL
April 2009 - July 2010
CCP4 Grant Renewal 2014-2019: Question-driven crystallographic data collection and advanced structure solution
BIOTECHNOLOGY & BIOLOGICAL SCIENCE RESEARCH COUNCIL
June 2015 - June 2019
Ab initio modelling for X-ray crystal structure solution
BIOTECHNOLOGY & BIOLOGICAL SCIENCE RESEARCH COUNCIL
August 2010 - July 2012
The structure-function relationship of anti-microbial peptides approached by ab initio protein modelling.
BRITISH SOCIETY FOR ANTIMICROBIAL CHEMOTHERAPY (UK)
June 2014 - August 2014
Development of lead compounds for trypanocidal drugs based on inhibitors targeted against parasite glycolysis (TRYGLYCHEMO).
EUROPEAN COMMISSION
November 2003 - October 2004
Exploring covariance-based models for protein crystal structure solution with AMPLE.
SCIENCE AND TECHNOLOGY FACILITIES COUNCIL
October 2014 - September 2018
The titin myofilament as emerging factor in cardiomyopathy
BRITISH HEART FOUNDATION (UK)
September 2013 - September 2016
Research Collaborations
Dr Ronan Keegan
External: STFC
Research collaboration
Dr Grzegorz Chojnowski
External: EMBL, Hamburg
Prof Jia Meng
External: XJTLU
Dr Kate Baker
Internal
Dr Andrew Carnell
Internal
Prof Pat Eyers
Internal